- セリウム同位体の(p, γ)反応実験及び陽子捕獲反応によるpプロセスのモデル計算
- 希薄フッ化水素酸中におけるラザホージウム(104Rf)のTTA吸着挙動を利用した陽イオン錯形成過程の研究
- 溶媒抽出法を用いた核種合成過程の異なるアスタチンの化学形の研究
- 液体シンチレーション測定を用いた海水中放射性ストロンチウム分析の迅速化
セリウム同位体の(p, γ)反応実験及び陽子捕獲反応によるpプロセスのモデル計算
Experimental Measurement of (p, γ) Reactions with Cerium Isotopes and Model Calculation of P-processes via Proton Capture Reaction
上野 慎吾
【序論】
太陽系中に存在する核種の内、鉄より重い核種のほとんどは中性子捕獲反応によって合成された。しかし中性子捕獲反応では合成されない陽子過剰核(p核)も少ない存在度ではあるが存在する。p核の生成経路(pプロセス)について、γ線による熱分解、ニュートリノとの核反応、陽子やヘリウム原子核との捕獲反応が考えられている。また元素合成にかかる時間にも短時間に生成されるモデルと100億年程度の時間をかけてゆっくりと生成されるモデルが考えられているが、いずれも同位体組成の定量的な説明には至っていない。その原因として、モデル構築に不可欠なp核合成に関わる核データが不足していることや考慮されていない反応の影響が挙げられる。またモデルを構築するとき放射性核種の反応も考慮しなければならず、理論による反応断面積の推定が必須となるが、その理論値の妥当性も問題となる。そこで新たな取り組みとして、遅い中性子捕獲反応(sプロセス)によってできる核種(s核)が陽子を捕獲するシナリオを考えp核の同位体組成の説明を試みた。今回着目している質量数130~150には天然に存在するp核が136Ce、138Ce、138La、144Smの4種類あり、年代法に利用される放射性p核146Smも存在している。天然に存在する4種のp核の存在量を説明可能なモデルを構築した上で146Smに適用すればpプロセスが起きた年代について議論できる。本研究では、s核への陽子捕獲反応によるpプロセスのモデル構築を行い、またp核である136Ce、138Ceの陽子捕獲反応断面積を取得することでモデルに用いた核データの再検討を行った。また実験によって得られた核反応断面積を考慮し補正をしたモデルにより、有効なpプロセスが起きる条件や年代について報告する。
【モデル計算】
初めにp核である136Ce、138Ce、138Laはs核のBa同位体、144Smはs核のNd同位体から合成されたと仮定しているため、純粋なsプロセスによるBa、Nd同位体比を次のように見積もった。同位体比は出発物質をそれぞれ133Cs、141Prとし、中性子密度4×108 n/cm3、4×108 Kの温度下でその飽和値を求めた。次にsプロセスで生成したBa、Nd同位体を出発物質として陽子捕獲反応を行い、136Ce、138Ce、138La、144Smの存在量の時間変化を次のように追った。中性子密度4×108 n/cm3、プロトン密度103~104 g/cm3、温度条件(0.5~2)×109 K、反応時間0秒~3000万年の範囲にてs核に対するp核の比及びp核同士の比、136, 138Ce/134Ba比、138La/134Ba比、138Ce/134Ba比、138Ce/138La比、136Ce/138Ce比、144Sm/142Nd比の時間変化を求め、全ての比が太陽系中の値と一致する条件(プロトン密度、温度、反応時間)を探した。また実験値を考慮した計算では、モデル計算に考慮した反応のうち陽子捕獲反応の断面積を全て変えたが、他の条件(プロトン密度、中性子密度、温度、反応時間)については固定した。モデル計算に用いた核反応断面積の値はNON-SMOKERコードによる理論値を採用した。
【実験】
プラズマスパッタリング法にて27Al箔(99.999%, 50 μm厚)にCe標的(136Ceまたは138Ceをそれぞれ濃縮したもの)を蒸着させた試料5枚、ビーム減衰用natTa箔(99.95%, 100 μm厚)2枚をスタックとし、大阪大学核物理研究センターのAVFサイクロトロンから供給される14 MeVの陽子ビームを1~4時間照射した。またCe標的1枚、natTa箔2枚、ビームモニター用natCu箔(99.9%, 10 μm厚)1枚のスタックに対して14 MeVの陽子ビームを30分照射した。照射終了後、各試料についてGe半導体検出器を用いたγ線スペクトロメトリーを行った。得られた核反応生成物の放射能から陽子捕獲反応断面積を求め、NON-SMOKERコードによる理論値の妥当性を評価した。
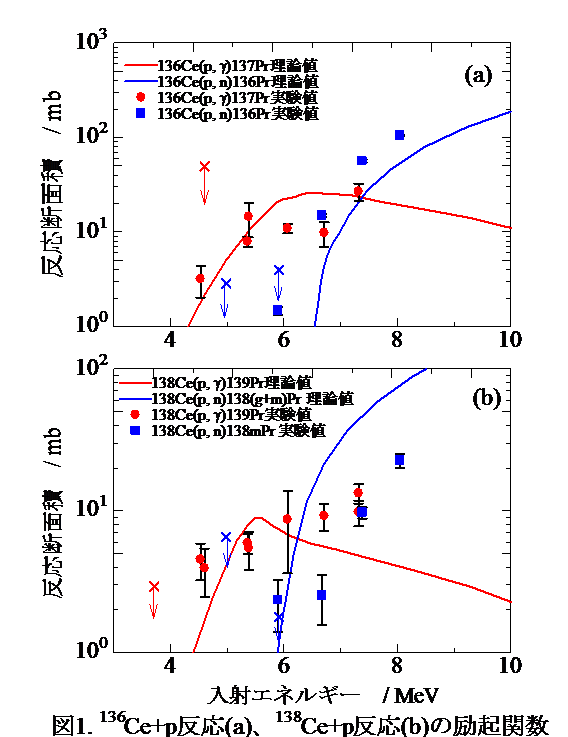
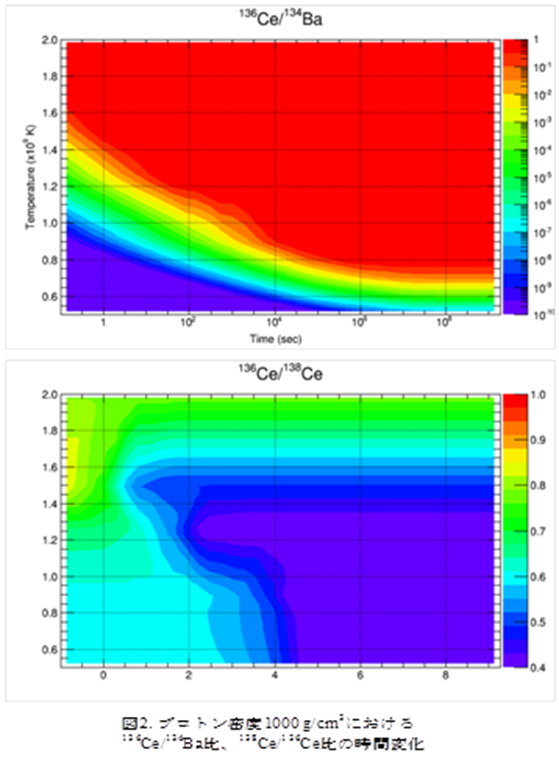
【結果と考察】
136, 138Ce(p, γ) 137, 139Pr反応、136, 138Ce(p, n) 136, 138mPr反応の生成断面積が初めて測定された。図1に136Ce+p反応及び138Ce+p反応の励起関数を示す。恒星内での核反応は主に低エネルギーの(p, γ)反応が起きているので、その反応だけに着目する。NON-SMOKERコードによる理論値と比較した場合、低エネルギー側(4~6 MeV)において理論値とほぼ同じ値(図1(a))から10倍大きい値(図1(b))の範囲にある。また、当研究室の林(144Sm+p系)やL. Netterdonら(130Ba+p系)についても同じズレの範囲内であると報告した。これより、NON-SMOKERコードによる陽子捕獲反応断面積の理論値は低エネルギー側において過小評価している可能性がある。
図2に実験値を考慮し陽子捕獲反応断面積を一律10倍に補正したモデル計算の結果の一部として、プロトン密度103/cm3の時の136Ce/134Ba比、136Ce/138Ce比の時間変化を表す等高線図を示す。横軸が反応時間、縦軸が温度、その時の色が同位体比を表し右側の縦棒に対応する。また、縦棒中の四角で囲まれた部分は太陽系中の値である。プロトン密度103/cm3、反応温度1.2~1.4×109 K、反応時間1~10 sの時に、今回着目している全ての同位体比が太陽系中の値と一致した。この結果から質量数130~150における現在の太陽系内のp核の存在量は、陽子捕獲反応シナリオにより説明可能であり、超新星爆発のような極めて短い時間スケールにて合成されることが示唆された。
希薄フッ化水素酸中におけるラザホージウム(104Rf)のTTA吸着挙動を利用した陽イオン錯形成過程の研究
Complex formation of a cationic rutherfordium (104Rf) fluoride in HF/HNO3 solutions studied by means of a TTA extraction chromatography
北山雄太
【序論】
超重元素 (Z≥104) は安定同位体が存在せず,化学実験に用いられる核種は通常半減期が数分以下と短寿命である。さらに製造手段である重イオン核反応による生成率も極めて小さい(例えば261Rfの場合 ~3原子/分 程度)。これらの特徴から,超重元素化学実験は単一原子化学としての取り扱いとなる。そのため,しばしば超重元素化学実験はこれに最適化した実験手法を用い,同族元素の化学挙動との比較によって実験を行い考察される。Ishiiらの陽イオン交換樹脂を用いたRf研究によればHF/HNO3溶液中においてRf及びその同族元素Zr,Hfは2価又は3価の陽イオンフッ化物錯体として存在すると報告されている1)。これに代表されるように,錯体配位子の濃度に依存した吸着挙動の研究により吸着錯体の価数や錯形成定数が明らかとなる。しかしながら,Rfのフッ化物錯形成定数の決定には至っていない。
荒木,武田らの先行研究2,3)では,Rfのフッ化物錯形成に関して理解を深めるため,キレート抽出剤であるTTAを抽出剤に用いて,逆相クロマトグラフィー用の樹脂を開発した。本研究では,先行研究にて開発したTTA樹脂を用いてHF/HNO3溶液系の希薄フッ酸濃度条件におけるRfの化学的性質を議論できる実験条件を,Zr,Hf無担体トレーサーを用いた実験により見出した。条件の検討には,最初にZr,Hfの抽出挙動を確認する目的で長半減期核種を用いたバッチ実験を行い,続いてRf実験の模擬実験として短半減期核種を用いてオンラインクロマトグラフィー実験を行った。これら結果を踏まえ,261Rfの吸着挙動の観測を行った。
【実験】
〔Zr,Hf実験〕
バッチ実験は,核反応により製造した88Zr,175Hf無担体トレーサーを用いて行った。一定質量のTTA樹脂と種々の濃度のHF/0.01 M HNO3を撹拌用チューブに分取し樹脂を調整した。Zr,Hfトレーサー溶液を加えて撹拌した。固液分離後,γスペクトロメトリーにより濾液中の核種を定量し,TTA樹脂への88Zr,175Hfの分配係数 (Kd) を求めた。一方,オンラインクロマトグラフィー実験ではγ線放出核種89mZr (T1/2 = 4.16 m)及び85Zr (T1/2 = 7.9 m),169Hf (T1/2 = 3.25 m)をそれぞれnatY(p, 2n)89mZr及びnatGe(18O, xn)85Zr, natGd(18O, xn)169Hf反応により合成して用いた。実験手順は下に示したRf実験と同様に行った。ここで溶離曲線を得られる流速条件を見出し,様々なHF/HNO3溶液を用いてKd値及び吸着率を求めた。
〔Rf実験〕
理化学研究所のK70 AVFサイクロトロンを用いて248Cm(18O, 5n)261Rf反応により104 261Rf (半減期68秒) を合成し,オンライン自動迅速化学分離装置 (ARCA)2)ならびに自動α線測定装置 (RIDER) を利用してTTAを抽出剤に用いた固定相に対する逆相クロマトグラフィーを行った。カラム (内径1.6 mm×7 mm) に充填する固定相は,50wt.%TTA-n-オクタノール溶液と保持担体 (三菱化学,CHP20/P20,粒径20 μm) を等重量比で混ぜ合わせて調製した。核反応生成物をKCl/Heガスジェット法 (流速2.5 L/min) により化学室に迅速搬送し,ARCAに約200秒間捕集した。その後,HF/0.01 M HNO3 (第一溶離液) を流速0.1 mL/minで85 μL流して,捕集物を溶液化しつつカラムに導入した。その後,0.1 M HF/0.1 M HNO3 (第二溶離液) を流速1.0 mL/minで 250 μL流し,すべての吸着イオンをカラムから溶出させた。Ta皿上に捕集したそれぞれの溶離液をハロゲンランプと熱したHeガスを用いて強熱することにより蒸発乾固し,RIDERのPIN検出器を用いてα線測定して吸着率を求めた。さらに,同時生成した169Hfをγ線スペクトロメトリーで定量し,収率と吸着率の変動をモニターした。
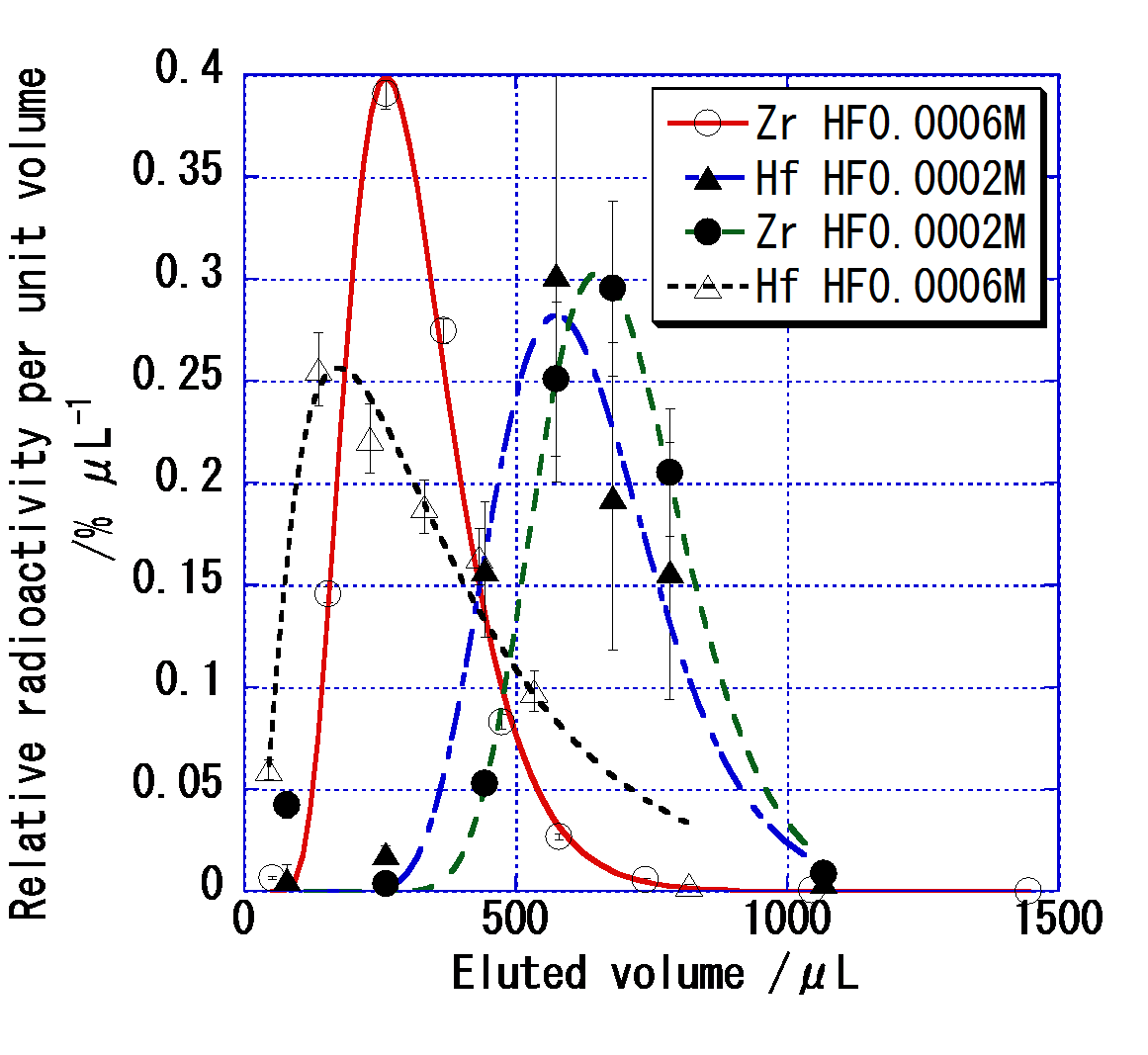
Fig. 1 Typical elution curves of Zr and Hf at a flow rate of 0.1 mL/min. The lines are drawn based on a theoretical model.
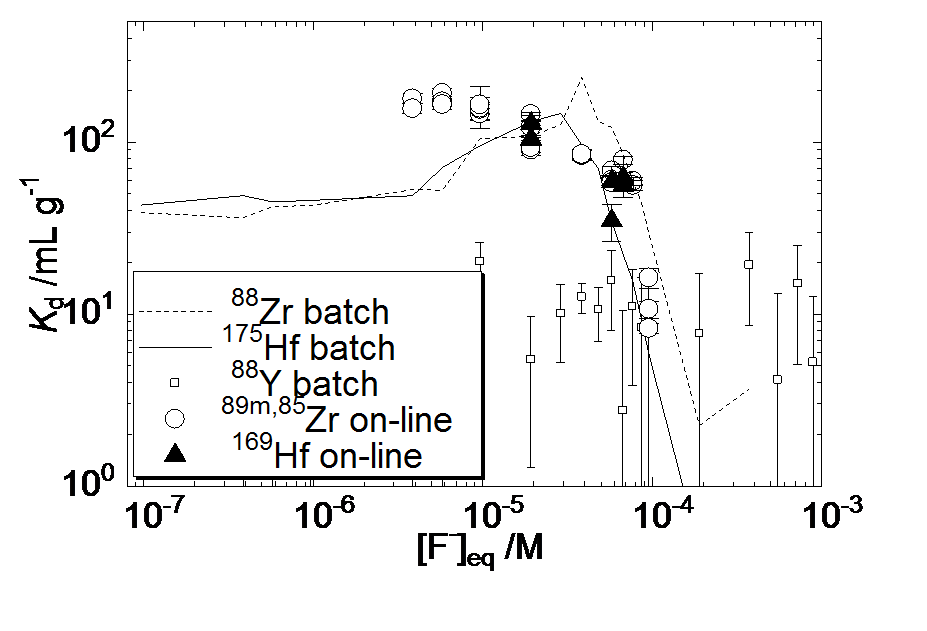 Fig. 2 Distribution coefficient (Kd) values by the batch and on-line experiments for 89m, 85Zr and 169Hf on the TTA resin as a function of the equilibrium concentration of F- ([F-]eq).
Fig. 2 Distribution coefficient (Kd) values by the batch and on-line experiments for 89m, 85Zr and 169Hf on the TTA resin as a function of the equilibrium concentration of F- ([F-]eq).
【結果と考察】
〔Zr,Hf実験〕
Fig. 1に流速0.1 mL/min時の典型的な溶離曲線を示す。1 mL/minまでの流速を用いて最適条件を検討し,0.1 mL/minにおいて様々な溶液の種類によりバッチ実験においてKd値を再現する条件を見出すことに成功した。次に,Fig. 2にバッチ実験及びオンライン実験から得たZr,Hfに対するKd値のフッ化物濃度([F-]eq)依存性を示す。両実験により得た結果はそれぞれよく再現しており,[F-]eqが高い領域においてはZr,Hf間の差が観測された。さらに[F-]eq> 4×10-5 Mでは,フッ化物イオン濃度の増加に従ってKd値が減少している。これは陰イオンフッ化物錯体が逐次形成されることで,陽イオン錯体が減少しているためと考えられる。これらにより [F-]eq>10-5 Mの条件がRf実験に適すると判断した。
〔Rf実験〕
Rfの溶離実験は合計で1001回行った。8.00-8.40 MeVのα粒子エネルギーの範囲内で同定した261Rf (8.28 MeV) 及びその娘核種257No (8.22,8.32 MeV) については,21回のα-α相関事象を含む160カウントが観測された。それぞれの条件に対して吸着率の値は第一,第二溶離液で観測された放射能から算出した。ただし,Rfの化学的挙動を反映しない257Noの寄与が考えられるため,シミュレーションによって影響を評価し実験値を補正した。Fig. 3にオンライン実験の結果として,[F-]eqに対するRf及びZr,Hfの吸着率の変化を示す。Rfの吸着率が[F-]eqに増加に伴って減少していることから,Rfが陽イオンフッ化物錯体としてTTAに抽出されており,Zr及びHfと同様にフッ化物錯体の逐次形成反応が起こっていることが明らかとなった。この結果は陽イオン交換樹脂に4族元素が2価又は3価の陽イオンフッ化物錯体を形成しているというIshiiらの主張と矛盾しない1)。さらに,これまでのイオン交換樹脂による実験結果では,Zr,Hfの挙動はほとんど一致していたが,本実験では両元素間に差が見られる興味深い結果を得た。[F-]eq>2×10-4 Mにおいて吸着率の順序はRf>(Zr≥)Hfであり,イオン半径の逆数の傾向と一致している4,5)。但し,Rfの正確な吸着率値の決定には257Noの溶離挙動の確認実験を行い,261Rfの吸着率を実験的に補正する必要がある。
![Fig. 3 Percent adsorption (%ads) values for <sup>85</sup>Zr, 169Hf and 261Rf on the TTA resin as a function of the [F-]eq.](superheavy1/superheavy1-3.png)
Fig. 3 Percent adsorption (%ads) values for 85Zr, 169Hf and 261Rf on the TTA resin as a function of the [F-]eq.
【参考文献】
- Y. Ishii et. al., Bull. Chem. Soc. Jpn. 84, No. 9, 903 (2011)
- 荒木幹生,金沢大学修士論文 2009年度
- 武田勇樹,金沢大学修士論文 2011年度
- C. D. Kacher et. al., Radiochim Acta 75, 135 (1996)
- R. D. Shannon, Acta Drystallogr., Sect. A 32, 751 (1976)
溶媒抽出法を用いた核種合成過程の異なるアスタチンの化学形の研究
Study of the Chemical Form of Astatine through the Several Nuclear Synthetic Processes by Means of Solvent Extraction
谷口 拓海
【序論】
がんの治療法として、放射性核種が標識された薬剤を人体に投与することで治療を行うアイソトープ治療がある。その応用が期待されている核種の一つである211Atについては、半減期が7.2時間と遠隔地での利用に対して制限がある。そこで本研究室では211Rn-211Atジェネレータ技術に着目してきた[1]。これは親核種となる211Rn(半減期:14.6時間)を製造し、その娘核種に当たる211Atをミルキングすることで211Atの使用制限範囲の拡大を可能にする。Atは他のハロゲン元素と比較して多くの酸化数を有し化学種が多いことが報告されている。これまで211At は209Bi(α,2n)211At反応によって直接的に合成されてきたが、本研究室で目指すジェネレータから供給される211Atは核種の合成過程が異なることから、合成直後の状態では異なる化学種となる可能性があり、その化学的挙動に対して比較検討が必要となる。しかし、Atは安定同位体を持たず、また反応性が高く微量の不純物の影響を受けやすいことから化学的挙動の解明が困難とされており、古くから溶媒抽出法やイオン交換法などの手法を用いた様々な検討が行われてきた[2,3]。本研究では、それらの手法の中でもBi標的からのAt分離法の一つであるジイソプロピルエーテル(DIPE)-塩酸による溶媒抽出法を選択し、核種合成過程の異なるAtの化学的挙動を比較することと、溶液中のAtの化学的挙動の解明を目的として、抽出プロセスにおけるAtの化学形を推定して抽出平衡式を提案する為の実験を行った。
【実験】
211Atはα線での定量が煩雑である為、直接的に209Bi(α,2n)211At反応によって製造する211Atの抽出挙動の確認の為には、類似の核反応である209Bi(α,3n)210At反応によって製造可能であり、定量に適したγ線放出核種である210Atを使用した。209Biを高純度Al箔(純度99.999%)に対して厚さ1.2-1.5 mg/cm2となるよう真空蒸着し、標的とした。この標的に対して加速器施設で照射実験を行い、209Bi(7Li,5n)211Rn反応と209Bi(α,3n)210At反応によってそれぞれ211Rnと210Atを製造した。製造した211Rnは211Atの壊変生成を待った後、また210Atは製造後そのまま溶液化を行った。溶液化した211Atと210Atを別々の塩酸溶液に加えて濃度調整し、等量の前平衡済みDIPEによって溶媒抽出を行い、抽出後の両相を分取して211Atに対しては液体シンチレーションカウンターによるα線測定、また210Atに対してはGe半導体検出器によるγ線測定を行うことで放射能濃度を決定し、分配比を算出した。また溶媒抽出における抽出機構を推定するために上記の実験に加えて、塩化物イオン濃度の影響を確認するために塩化ナトリウムを添加する実験と、またAtの化学形を特定の酸化状態に統一して考察するために酸化剤、または還元剤を添加する実験を行った。これら追加の実験には直接的な核反応によって製造した210Atをトレーサーとして使用した。酸化剤としては硝酸、還元剤としてはアスコルビン酸を選択し、それぞれ溶液中におけるAtの化学種を酸化状態ではAtx+、還元状態ではAt-となっていると仮定して抽出平衡式を提案することとした。
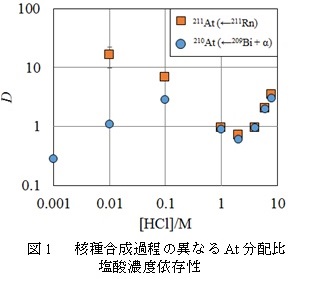
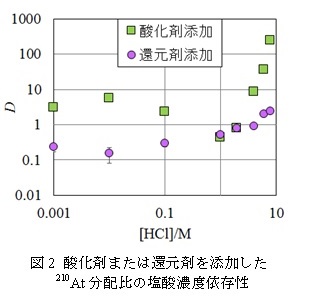
【結果・考察】
211Rn-211Atジェネレータからミルキングすることで得た211Atと直接的に核反応によって製造した210Atの塩酸濃度に対する分配比の依存性を図1に示した。塩酸濃度1 M以上では二つの抽出挙動は一致した。核種合成過程の異なるアスタチンが、塩酸濃度1 M以上においては同一の化学的挙動を取ることを確認した。
さらにその抽出機構を解明するために行った追加の実験について以下に示す。まず酸化剤または還元剤を添加した210At分配比の塩酸濃度依存性の結果を図2に示した。還元剤を添加したものは塩酸濃度に依存して一定の傾きで分配比が上昇する一方、酸化剤を添加したものは異なる結果となった。また塩化ナトリウムを添加して塩化物イオン濃度を変化させる実験を行ったところ、酸化剤を添加したものでは塩化物イオン濃度の変化に対して分配比が影響を受ける様子が確認されたが、還元剤を添加した分配においては塩化物イオン濃度に対する依存性が確認されなかった。
この結果から、酸化剤または還元剤を添加した溶液中のAtの化学形をそれぞれAtx+、At-と仮定することで抽出機構の平衡式を以下のように提案した。酸化化学種については過去の報告と一致した結果となり[2]、また還元化学種については本研究により新たに提案された。ただし、塩酸濃度1M以下ではこれ以外の他の抽出機構の影響が示唆される結果となった。
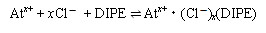 :(酸化化学種の平衡式)
:(酸化化学種の平衡式)
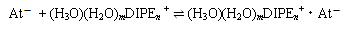 :(還元化学種の平衡式)
:(還元化学種の平衡式)
本研究の結果より、211Rn-211Atジェネレータからミルキングされる211Atの化学形は、従来法である直接的な核反応による211Atの化学形とは必ずしも一致せず、酸濃度の調整によって同一の状態にすることができることがわかった。
【参考文献】
- 前田英太, 金沢大学大学院修士論文(2013)
- Alliot, C. et al, Radiochimica. Acta. 97, 161-165 (2009)
- Roy, K., Lahiri, S., Applied Radiation and Isotopes 66, 571-576 (2008)
液体シンチレーション測定を用いた海水中放射性ストロンチウム分析の迅速化
Rapid analysis of radiostrontium in seawater through the use of liquid scintillation methods
渡邊 良祐
【序論】
ストロンチウム90 (90Sr) は先の福島第一原子力発電所事故において、陸域への影響は少なかったが、冷却水に溶解したものが汚染水となり、地下への漏えいと海洋への放出が続いている[1]。現在、海水中の90Srの分析には文部科学省の放射能測定法(発煙硝酸法)[2]が用いられているが、危険な上に手順が多く迅速分析という点においては改善の余地がある。そこで本研究では、新しい化学分離スキームの構築と測定法の最適化を目的とした。化学分離では、クエン酸存在下でリン酸塩沈殿分離をすることによって海水中の共存イオンを除去し、海水には適用できないとされてきたSr Rad Disk[3]の適用を試みた。また、測定法の最適化として液体シンチレーション測定(LSC)により、抽出シンチレータを用いて90Srを抽出し、そのβ線を直接測定する方法を検討した。
【実験】
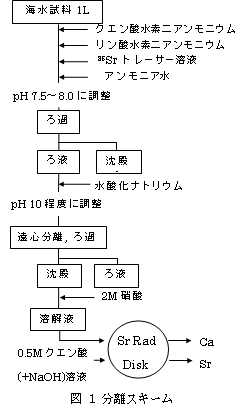
(a) Srの濃縮分離
Srを選択的に捕集できるSr Rad Diskを海水に適用するため、リン酸塩沈殿分離によりCa, SrをK, Na, Mgなどから分離することを試みた。この際、クエン酸添加とpH変化によるSrの挙動を観察した。回収率の計算には85Srトレーサー(γ線 514keV)を用いた。
(b) Srの精製
沈殿分離により大部分の共存イオンを除去できたので、次にCaを除去するためにSr Rad DiskによりSrを精製した。加えて、DiskからのSrの脱離における最適条件を検討した。
(c) LSCによるβ線測定
抽出シンチレータによって90Sr-90Yを有機相に抽出して測定する二相法の検討を行った。また、乳化シンチレータ法及びチェレンコフ光測定法と比較した。また、測定試料の調製に際し、Sr抽出におけるクエン酸溶液の濃度とpH依存性を確認した。
(d) 海水試料への適用
図1の条件を最適化した分離スキームを用いて、福島原発近傍の海水試料とIAEA-PT試料(IAEAから提供された標準試料) の分析を試みた。
【結果と考察】
(a) 海水試料にクエン酸を添加することで、沈殿分離の際にSrの損失を抑えてMgを除去することができ、K, Naは沈殿しないで除去された。また、Mg沈殿によるSrの損失を抑えるには再沈殿が有効であることも分かった。
(b) 海水にSr Rad diskは適用できないとされていたが、共存塩を除去することで適用可能となった。また、DiskからSrを脱離する際に一般的に用いられるEDTA溶液の代わりに、0.5 Mクエン酸溶液に少量の水酸化ナトリウム溶液を加えることで、EDTAと同等の効果を得た。
(c) 各測定法の測定条件を表1に示す。特に二相法を用いた場合、低バックグラウンドと高い計数効率で90Srを測定でき、クエン酸溶液からpH5~7で高い抽出率が得られた。しかしpH>8程度に上げた場合にはシンチレータが白濁した。また、硝酸を用いてpH<4に調整した場合はSrがほとんど抽出されずYのみが抽出されることが分かった。
| 測定核種 | 計数効率 (%) | BG. (cps) | |
|---|---|---|---|
| 乳化シンチレータ法 | 90Sr + 90Y | 92 | 2.5 |
| 二相法 (pH=2) | 90Y | 98 | 0.27 |
| 二相法 (pH=11) | 90Sr + 90Y | 96 | |
| チェレンコフ光測定 | 90Y | 55 | 0.14 |
(d) 海水、IAEA-PT試料に適用した結果を表2に示す。
| 試料 | 放射能 |
|---|---|
| 福島海水 | 0.10 ± 0.01 (Bq/L) |
| IAEA-PT (Sample01) | 20.9 ± 3.2 (Bq/kg) |
【まとめ】
Diskの交換容量の制限から海水の試料量は1 L以下に制限されたが、図1の分離スキームによる85Srの回収率は84%が得られ、分離時間は約3.5 時間と短時間での分離が可能となった。 液体シンチレーション測定において、抽出シンチレータを用いた 90Sr・90Y弁別測定が可能になった。これは将来的に、緊急時モニタリングにおいて重要とされる、89Srを含めた放射性ストロンチウムの定量への発展が期待される。
【参考文献】
- 原子力規制委員会 (http://radioactivity.nsr.go.jp/ja/index.html)
- 文部科学省「放射能測定法シリーズ」No.2:放射性ストロンチウム分析法(昭和58年3訂)
- 住友スリーエム HP (http://www.mmm.co.jp/filter/empore/rad/pdf/sced11-011.pdf)